















Pompe disease is a hereditary disorder which is often caused due to defect in the genome of the human body. This disease is formed when glycogen is formulated and accumulated in the cell of a human body. Although a rare form of disease, Pompe is considered as a fatal disease of the neuromuscular system.
History of Pompe Disease
This disease was first discovered in an infant in the year of 1932. Since this disease was first discovered by Dr. Johannes C. Pompe, it was named after him.
Rarity of the Pompe Disease & its cause
This disease is known to be a rare one because 1 infant out of 40000 is affected. The reason why pompe disease occurs is because GAA i.e. acid alpha glucosidase is deficient in the infant. This enzyme is required to break the glycogen present in the body. Since the glycogen present in the body does not break, it keeps on getting accumulating, particularly in the skeletal system and heart.
3 kinds of Pompe Disease
After years of study, the researchers have found out 3 kinds of Pompe disease which are described as below:
- Classic In – fantile onset
- Non – classical infantile onset
- Non – classical infantile late onset
Classic in – fantile onset
It is usually seen in infants within a few months after they are born. The infants experience weakness of the muscles. Other symptoms of this disorder are heart defects, breathing problem and enlarged liver.
Symptoms:
- Difficulty in feeding
- Difficulty in hearing
- Feeling distress during normal respiratory activity
Non – classical infantile onset
Second form of disease i.e. the non – classical occurs when the infant reaches the age of one. Children are not able to develop their muscles completely and hence they suffer from major respiratory distress during breathing. These infants also have an enlarged heart and also they are able to pass only a few years of the childhood.
Non – classical infantile late onset
This form of Pompe disease is seen in adolescents or teenagers. Muscle weakness is most prominently seen in the legs as well as in the trunk.
Symptoms:
- Sleep apnea
- Facing difficulty in breathing
- Consistent weakness of the muscles
- Liver enlargement
- Hyperlordosis
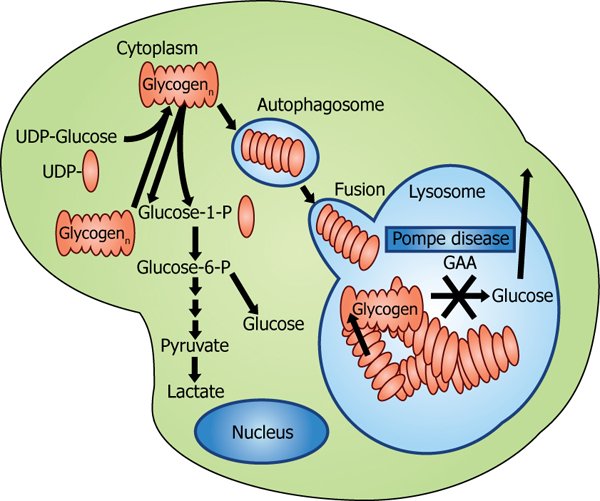
Diagnosis of Pompe Disease
ECG i.e. electro cardio gram and Chest X – ray can be used for diagnosis of pompe disease. Muscle tissue biopsy is also performed which allows to detect the glycogen content and muscle acid alpha glucosidase.
Treatment for the Pompe Disease
FDA i.e. the Food and Drugs Administration has approved a drug, named Myzozyme for the treatment of the pompe disease for children and infants. In case of late – onset, a drug named, Lumizyme has been approved. The treatment for the pompe disease works on the following understanding:
Since the enzyme GAA, as described above is missing or prevalent in deficient quantities in the infant, enzyme replacement therapy is most popularly used. This therapy helps in maintaining the heart size of the infant and also enhancing the muscle functioning of the infant or the child.







